What role does ADAMTS13 play in TTP?
5 min
Structure and function of ADAMTS13
ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin motifs 13) is a constitutively active enzyme (plasma metalloprotease) that catalyzes the breakdown of ultra-large and high molecular weight von Willebrand factor (VWF) into smaller multimers, reducing their thrombogenic potential, and maintaining hemostasis.1,2
ADAMTS13 is synthesized and secreted by endothelial cells, megakaryocytes, and hepatic stellate cells into the blood.2-4 ADAMTS13’s plasma half-life is 2–3 days.2
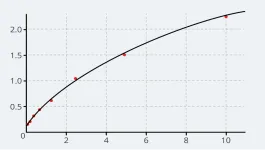
ADAMTS13 is known to cleave only one substrate, VWF, and does so at only one site in it’s a2 domain between Tyr1605 and Met1606.2
The activity of ADAMTS13 is dynamically regulated by conformational changes (unfolding) of the VWF molecule when it is exposed to the shear force of blood flow.2
If there is an ADAMTS13 deficiency thrombogenic VWF multimers persist in vessels where they bind platelets, leading to VWF/platelet aggregates and thrombosis in the microvasculature of the brain, heart, and kidney.2
ADAMTS13 deficiency may be due to genetic mutations or autoantibodies neutralizing its activity.2
From a screen of the large-scale sequencing data gnomAD database, 2,212 variants of ADAMTS13 were identified, of which 250 were pathogenic.5












Share this article with a colleague
Share this article with a colleague
This website is intended for an international audience of healthcare professionals outside of the US & UK. It should not be shared with patients, carers or the general public.
Abbreviations, Glossary and References
Abbreviations
ADAMTS13; A disintegrin and metalloproteinase with a thrombospondin motifs 13
gnomAD; Genome aggregation database
TTP; Thrombotic thrombocytopenic purpura
VWF; Von Willebrand factor
WPB; Weibel-Palade body
Glossary
ADAMTS13; ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin motifs 13) is a constitutively active enzyme (plasma metalloprotease) that catalyzes the breakdown of ultra large and high molecular weight von Willebrand factor (VWF) into smaller multimers, reducing their thrombogenic potential, and maintaining hemostasis.1,2
Incidence; The rate of new cases or events over a specified period for the population at risk for a certain event.
Microangiopathic hemolytic anemia (MAHA); Process of red blood cell destruction within the microvasculature accompanied by thrombocytopenia due to platelet activation and consumption. Thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) are primary forms of thrombotic microangiopathies.13
Prevalence; The proportion of a particular population found to be affected by a medical condition at a specific time.
Schistocyte; Circulating fragments of red blood cells commonly seen in blood smears from patients with thrombotic microangiopathies including TTP.14
Thrombocytopenia; Refers to a state of reduced peripheral platelets below normal levels (150x109/L) and can be caused by a wide variety of aetiologies that either decrease platelet production or increase platelet consumption.15
Thrombotic microangiopathy (TMA); TMA includes a diverse set of syndromes that can be hereditary or acquired, which can occur in children and adults with sudden or gradual onset.
TMA syndromes, despite being diverse, have a common set of clinical and pathological features: MAHA, thrombocytopenia, organ injury, vascular damage manifested by arteriolar and capillary thrombosis with characteristic abnormalities in the endothelium and vessel wall.16
Thrombotic thrombocytopenic purpura (TTP); TTP is a type of MAHA presenting with moderate or severe thrombocytopenia. There is associated organ dysfunction, including neurologic, cardiac, gastrointestinal and renal involvement; oliguria or anuric renal failure requiring renal replacement therapy is not typically a feature. TTP is confirmed by a severe deficiency (<10%) of ADAMTS13 activity.17
von Willebrand factor (VWF); VWF plays two key roles in hemostasis: 1) in primary (platelet-mediated) hemostasis, VWF binds to collagen and platelets thus promoting platelet activation and aggregation, and 2) in secondary (coagulation factor mediated) hemostasis VWF binds factor VIII (FVIII) protecting FVIII from rapid clearance. When VWF binds to collagen following vascular injury, it releases FVIII, leading to FVIII activation and initiation of the coagulation cascade.10,18
References
- Markham-Lee, Z., N.V. Morgan, and J. Emsley, Inherited ADAMTS13 mutations associated with Thrombotic Thrombocytopenic Purpura: a short review and update. Platelets, 2023. 34(1): p. 2138306.
- Kremer Hovinga, J.A., et al., Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers, 2017. 3: p. 17020.
- Suzuki, M., et al., Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem Biophys Res Commun, 2004. 313(1): p. 212-216.
- Zheng, X.L., ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med, 2015. 66: p. 211-225.
- Seidizadeh, O., et al., Estimating the population-based prevalence of congenital TTP using large-scale genetic databases. 12th BIC International Conference, 2023. 29(S3): p. 7.
- Crawley, J.T., et al., Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood, 2011. 118(12): p. 3212-3221.
- De Ceunynck, K., S.F. De Meyer, and K. Vanhoorelbeke, Unwinding the von Willebrand factor strings puzzle. Blood, 2013. 121(2): p. 270-277.
- Ruggeri, Z.M., The role of von Willebrand factor in thrombus formation. Thromb Res, 2007. 120 Suppl 1(Suppl 1): p. S5-9.
- Scully, M., et al., Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol, 2012. 158(3): p. 323-335.
- Stockschlaeder, M., R. Schneppenheim, and U. Budde, Update on von Willebrand factor multimers: focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul Fibrinolysis, 2014. 25(3): p. 206-216.
- Bryckaert, M., et al., Of von Willebrand factor and platelets. Cell Mol Life Sci, 2015. 72(2): p. 307-326.
- Leebeek, F.W. and J.C. Eikenboom, Von Willebrand's Disease. N Engl J Med, 2016. 375(21): p. 2067-2080.
- Arnold, D.M., C.J. Patriquin, and I. Nazy, Thrombotic microangiopathies: a general approach to diagnosis and management. CMAJ, 2017. 189(4): p. E153-E159.
- Zini, G., et al., ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes. Int J Lab Hematol, 2012. 34(2): p. 107-116.
- Gauer, R.L. and M.M. Braun, Thrombocytopenia. Am Fam Physician, 2012. 85(6): p. 612-622.
- George, J.N. and C.M. Nester, Syndromes of thrombotic microangiopathy. N Engl J Med, 2014. 371(7): p. 654-666.
- Scully, M., et al., Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost, 2017. 15(2): p. 312-322.
- Rauch, A., et al., On the versatility of von Willebrand factor. Mediterr J Hematol Infect Dis, 2013. 5(1): p. e2013046.